I45.81
Long QT syndrome (LQTS) is a condition which affects
repolarization of the heart after a heartbeat. It results in an increased risk
of an irregular heartbeat which can result in palpitations, fainting, drowning,
or sudden death. These episodes can be triggered by exercise or stress.Other
associated symptoms may include hearing loss.
Long QT syndrome may be present at birth or develop later
in life.The inherited form may occur by itself or as part of larger genetic
disorder. Onset later in life may result from certain medications, low blood
potassium, low blood calcium, or heart failure. Medications that are implicated
include certain antiarrhythmic, antibiotics, and antipsychotics. Diagnosis is
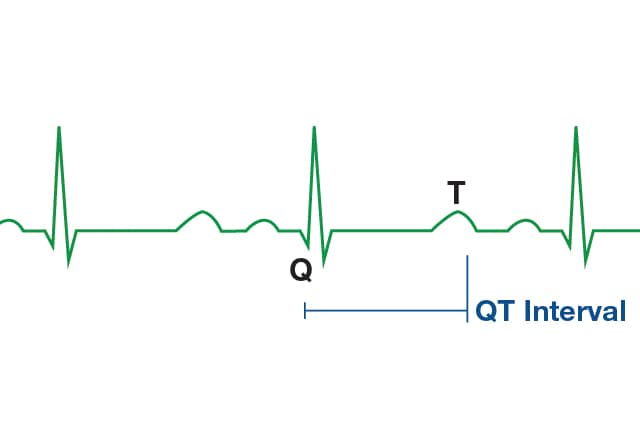
based on an electrocardiogram (EKG) finding a corrected QT interval of greater
than 440 to 500 milliseconds together with clinical findings.
Overview
Long QT syndrome (LQTS)
is a heart rhythm condition that can potentially cause fast, chaotic
heartbeats. These rapid heartbeats might trigger a sudden fainting spell or
seizure. In some cases, the heart can beat erratically for so long that it
causes sudden death.
Long
QT syndrome is treatable.
In
some cases, treatment for long QT syndrome involves surgery or an implantable
device.
Symptoms
Many people who have long
QT syndrome don't have any signs or symptoms. You might be aware of your
condition only because of:
· Results of an electrocardiogram (ECG)
done for an unrelated reason
· A family history of long QT syndrome
· Genetic testing results
For people who do experience signs and symptoms of long QT syndrome, the most common include:
· Fainting. This is the most common sign of long QT syndrome. Long QT
syndrome-triggered fainting spells (syncope) are caused by the heart
temporarily beating in an erratic way.
· Seizures. If the heart continues to beat erratically, the
brain will eventually not get enough oxygen, which can cause seizures.
· Sudden death. Generally, the heart returns
to its normal rhythm. If this doesn't happen by itself, or if an external
defibrillator isn't used in time to convert the rhythm back to normal, sudden
death will occur.
Inherited long QT syndrome
At least 17 genes
associated with long QT syndrome have been found so far, and hundreds of
mutations within these genes have been identified. Mutations in three of these
genes account for about 75 percent of long QT syndrome cases, while mutations
in the other minor genes contribute a small percent of long QT syndrome cases.
About 20 percent of
people who definitely have congenital long QT syndrome have a negative genetic
test result. On the other hand, among families with genetically established
long QT syndrome, between 10 percent and 37 percent of the relatives with a
positive long QT syndrome genetic test have a normal QT interval.
Doctors have described
two forms of inherited long QT syndrome:
· Romano-Ward
syndrome. This more common form occurs in people who
inherit only a single genetic variant from one parent.
· Jervell
and Lange-Nielsen syndrome. This rare form usually occurs
earlier and is more severe. In this syndrome, children inherit genetic variants
from both parents. They have long QT syndrome and also are born deaf.
Additionally, scientists
have been investigating a possible link between sudden infant death syndrome
(SIDS) and long QT syndrome and have discovered that approximately five to 10
percent of babies affected by SIDS had a genetic defect or mutation for long QT
syndrome.
Acquired long QT syndrome
Acquired long QT syndrome
can be caused by certain medications, electrolyte abnormalities such as low
body potassium (hypokalemia) or medical conditions. More than 100 medications —
many of them common — can lengthen the QT interval in otherwise healthy people
and cause a form of acquired long QT syndrome known as drug-induced long QT
syndrome.
Medications that can
lengthen the QT interval and upset heart rhythm include:
· Certain antibiotics
· Certain antidepressant and antipsychotic
medications
· Some antihistamines
· Diuretics
· Medications used to maintain normal
heart rhythms (antiarrhythmic medications)
· Some anti-nausea medications
Risk
factors
People who may have a
higher risk of inherited or acquired long QT syndrome may include:
· Children, teenagers and young adults
with unexplained fainting, unexplained near drownings or other accidents,
unexplained seizures, or a history of cardiac arrest
· Family members of children, teenagers
and young adults with unexplained fainting, unexplained near drownings or other
accidents, unexplained seizures, or a history of cardiac arrest
· First-degree relatives of people with
known long QT syndrome
· People taking medications known to cause
prolonged QT intervals
· People with low potassium, magnesium or
calcium blood levels — such as those with the eating disorder anorexia nervosa
Inherited long QT
syndrome often goes undiagnosed or is misdiagnosed as a seizure disorder, such
as epilepsy. However, long QT syndrome might be responsible for some otherwise
unexplained deaths in children and young adults. For example, an unexplained
drowning of a young person might be the first clue to inherited long QT
syndrome in a family.
Complications
Most of the time,
prolonged QT intervals in people with long QT syndrome never cause problems.
However, physical or emotional stress might "trip up" a heart that is
sensitive to prolonged QT intervals. This can cause the heart's rhythm to spin
out of control, triggering life-threatening, irregular heart rhythms (arrhythmias)
including:
· Torsades de pointes —
'twisting of the points.'
· Ventricular fibrillation.
Short
QT syndrome
Short QT syndrome is a condition that can cause a disruption of the heart's normal rhythm (arrhythmia). In people with this condition, the heart (cardiac) muscle takes less time than usual to recharge between beats. The term "short QT" refers to a specific pattern of heart activity that is detected with an electrocardiogram (EKG), which is a test used to measure the electrical activity of the heart. In people with this condition, the part of the heartbeat known as the QT interval is abnormally short. If untreated, the arrhythmia associated with short QT syndrome can lead to a variety of signs and symptoms, from dizziness and fainting (syncope) to cardiac arrest and sudden death. These signs and symptoms can occur any time from early infancy to old age. This condition may explain some cases of sudden infant death syndrome (SIDS), which is a major cause of unexplained death in babies younger than 1 year. However, some people with short QT syndrome never experience any health problems associated with the condition.
Causes Mutations in the
KCNH2, KCNJ2, and KCNQ1 genes can cause short QT syndrome. These genes provide
instructions for making channels that transport positively charged atoms (ions)
of potassium out of cells. In cardiac muscle, these ion channels play critical
roles in maintaining the heart's normal rhythm. Mutations in the KCNH2, KCNJ2,
or KCNQ1 gene increase the activity of the channels, which enhances the flow of
potassium ions across the membrane of cardiac muscle cells. This change in ion
transport alters the electrical activity of the heart and can lead to the
abnormal heart rhythms characteristic of short QT syndrome. Some affected individuals
do not have an identified mutation in the KCNH2, KCNJ2, or KCNQ1 gene. Changes
in other genes that have not been identified may cause the disorder in these
cases.
Diagnosis
Short QT syndrome is diagnosed
primarily using an electrocardiogram (ECG), but may also take into account the
clinical history, family history, and possibly genetic testing. Whilst a
diagnostic scoring system has been proposed that incorporate all of these
factors (the Gollob score), it is uncertain whether this score is useful for
diagnosis or risk stratification, and the Gollob score has not been universally
accepted by international consensus guidelines. There continues to be
uncertainty regarding the precise QT interval cutoff that is should be used for
diagnosis.
12-lead ECG (or KOLIBRI)
The mainstay of diagnosis of short QT syndrome is the 12-lead ECG. The
precise QT duration used to diagnose the condition remains controversial with
consensus guidelines giving cutoffs varying from 330 ms, 340 ms or even 360 ms
when other clinical, familial, or genetic factors are present. The QT interval
normally varies with heart rate, but this variation occurs to a lesser extent
in those with short QT syndrome. It is therefore recommended that the QT
interval is assessed at heart rates close to 60 beats per minute. Other
features that may be seen on the ECG in short QT syndrome include tall, peaked
T-waves and PR segment depression.
Published on 23 March 2019